Commits on Source (269)
- 169 additional commits have been omitted to prevent performance issues.
Showing
- .gitignore 9 additions, 0 deletions.gitignore
- .gitlab-ci.yml 42 additions, 21 deletions.gitlab-ci.yml
- .gitlab/merge_request_templates/merge_request.md 12 additions, 0 deletions.gitlab/merge_request_templates/merge_request.md
- CHANGELOG.md 57 additions, 0 deletionsCHANGELOG.md
- LICENSE.md 15 additions, 0 deletionsLICENSE.md
- README.md 36 additions, 28 deletionsREADME.md
- astrocyte_pkg.yml 15 additions, 9 deletionsastrocyte_pkg.yml
- docs/flowchart.pdf 0 additions, 0 deletionsdocs/flowchart.pdf
- docs/images/phantompeakqualtools.png 0 additions, 0 deletionsdocs/images/phantompeakqualtools.png
- docs/index.md 27 additions, 21 deletionsdocs/index.md
- docs/phantompeaks.md 10 additions, 0 deletionsdocs/phantompeaks.md
- docs/references.md 5 additions, 2 deletionsdocs/references.md
- docs/references.txt 0 additions, 49 deletionsdocs/references.txt
- test_data/A_1.bedpe.gz 0 additions, 0 deletionstest_data/A_1.bedpe.gz
- test_data/A_1.bedse.gz 0 additions, 0 deletionstest_data/A_1.bedse.gz
- test_data/A_1.tagAlign.gz 0 additions, 0 deletionstest_data/A_1.tagAlign.gz
- test_data/B_1.bedpe.gz 0 additions, 0 deletionstest_data/B_1.bedpe.gz
- test_data/B_1.bedse.gz 0 additions, 0 deletionstest_data/B_1.bedse.gz
- test_data/B_1.tagAlign.gz 0 additions, 0 deletionstest_data/B_1.tagAlign.gz
- test_data/design_single_contol_SE.txt 3 additions, 0 deletionstest_data/design_single_contol_SE.txt
CHANGELOG.md
0 → 100644
LICENSE.md
0 → 100644
No preview for this file type
docs/images/phantompeakqualtools.png
0 → 100644
{kind=link}
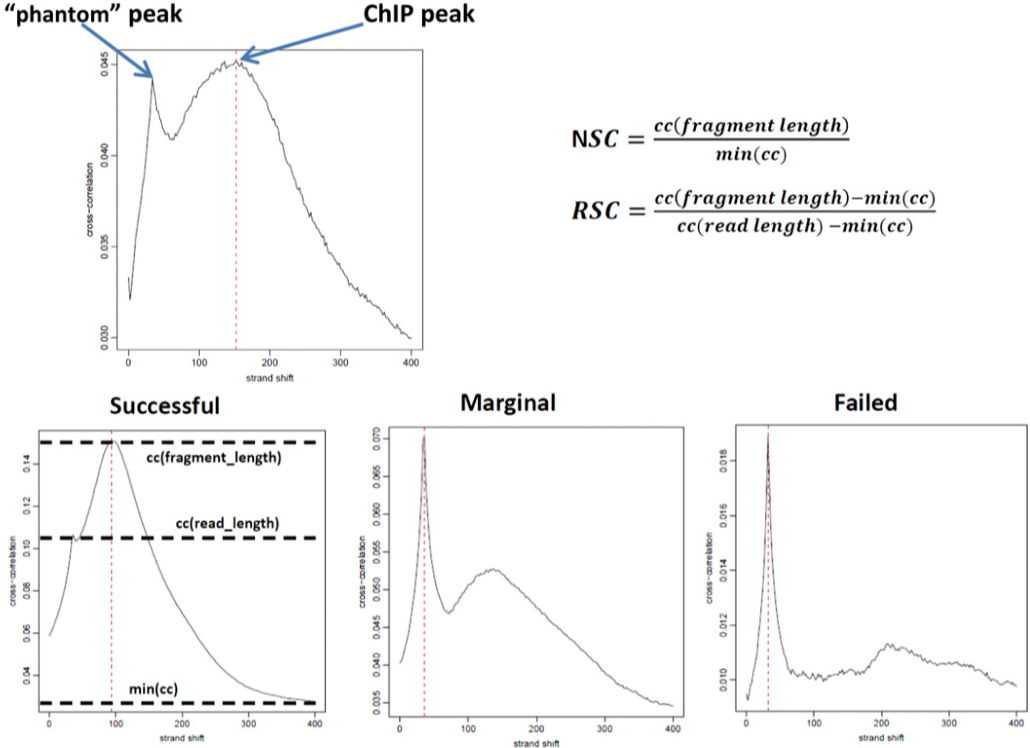
247 KiB
docs/phantompeaks.md
0 → 100644
docs/references.txt
deleted
100644 → 0
test_data/A_1.bedpe.gz
0 → 100644
File added
test_data/A_1.bedse.gz
0 → 100644
File added
test_data/A_1.tagAlign.gz
0 → 100644
File added
test_data/B_1.bedpe.gz
0 → 100644
File added
test_data/B_1.bedse.gz
0 → 100644
File added
test_data/B_1.tagAlign.gz
0 → 100644
File added
test_data/design_single_contol_SE.txt
0 → 100644