Merge branch '6-processCounts' into 'develop'
Resolve "process_count" See merge request !22
Showing
- .gitlab-ci.yml 9 additions, 0 deletions.gitlab-ci.yml
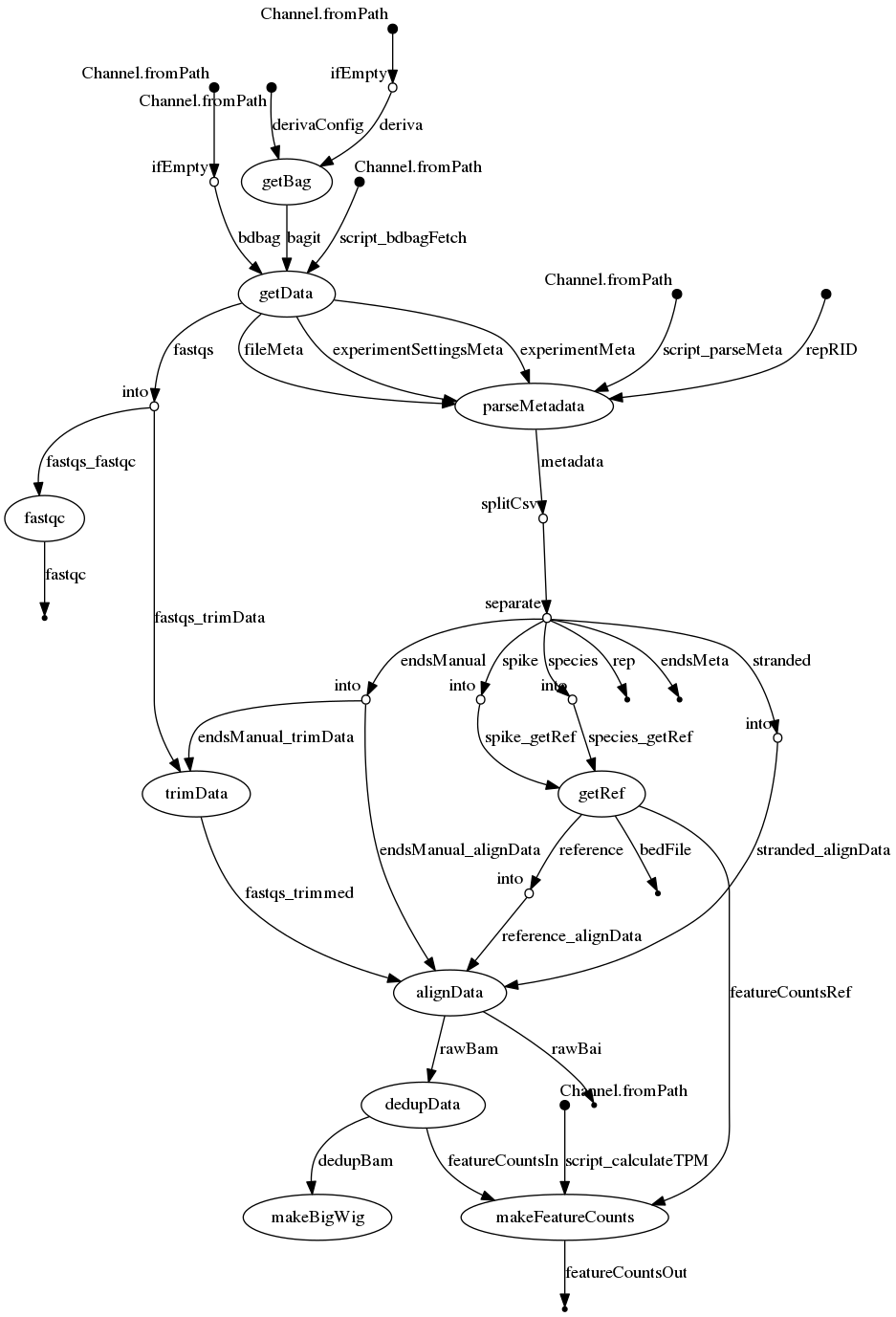
- dag.png 0 additions, 0 deletionsdag.png
- workflow/nextflow.config 3 additions, 0 deletionsworkflow/nextflow.config
- workflow/rna-seq.nf 190 additions, 96 deletionsworkflow/rna-seq.nf
- workflow/scripts/calculateTPM.R 30 additions, 0 deletionsworkflow/scripts/calculateTPM.R
- workflow/tests/test_makeFeatureCounts.py 15 additions, 0 deletionsworkflow/tests/test_makeFeatureCounts.py
dag.png
0 → 100644
{kind=link}
157 KiB
workflow/scripts/calculateTPM.R
0 → 100644
workflow/tests/test_makeFeatureCounts.py
0 → 100644