Merge branch 'new.qc.out' into 'develop'
Metadata output update Closes #56 and #52 See merge request !36
Showing
- .gitignore 2 additions, 0 deletions.gitignore
- .gitlab-ci.yml 6 additions, 2 deletions.gitlab-ci.yml
- README.md 8 additions, 2 deletionsREADME.md
- cleanup.sh 2 additions, 0 deletionscleanup.sh
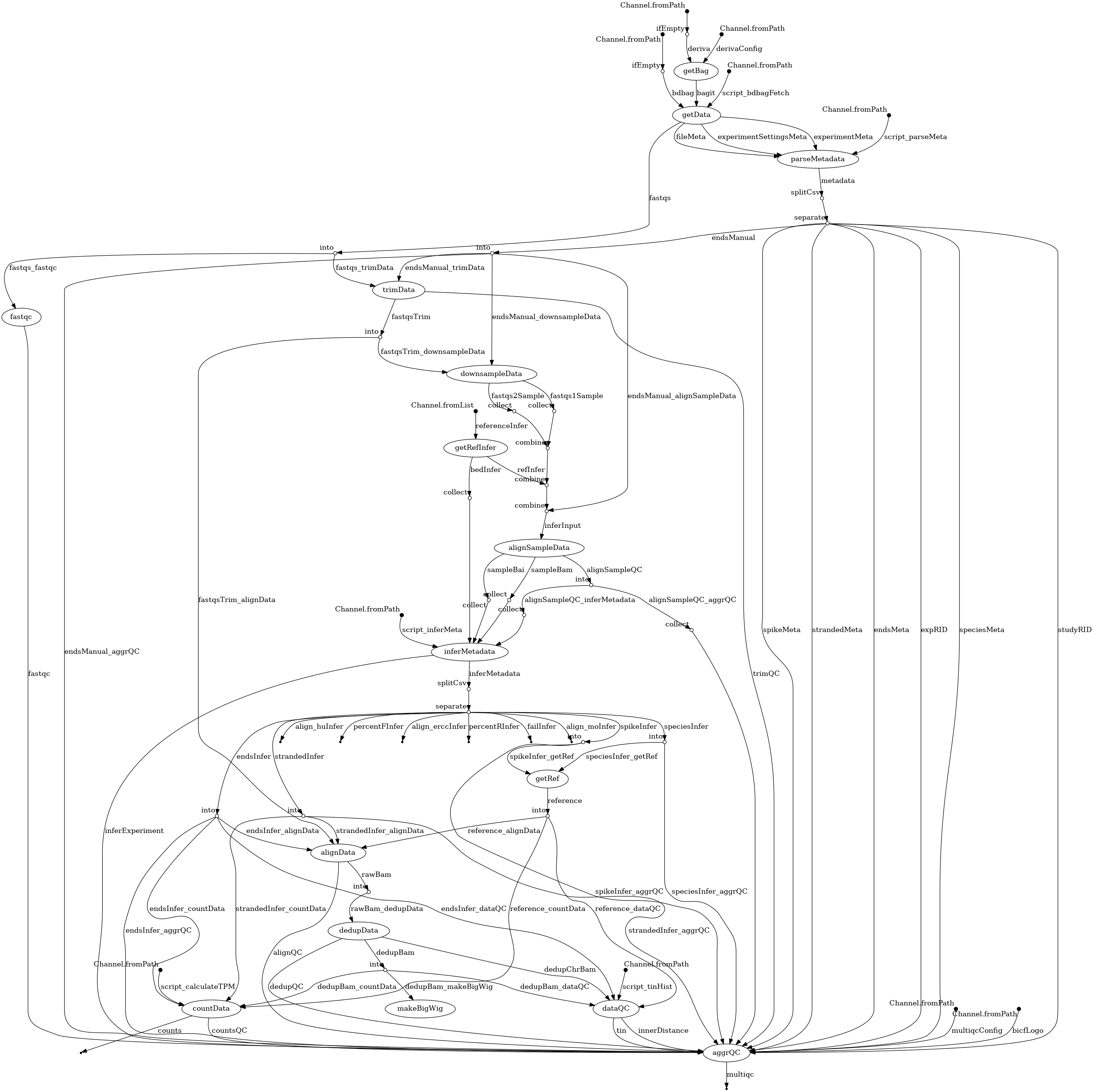
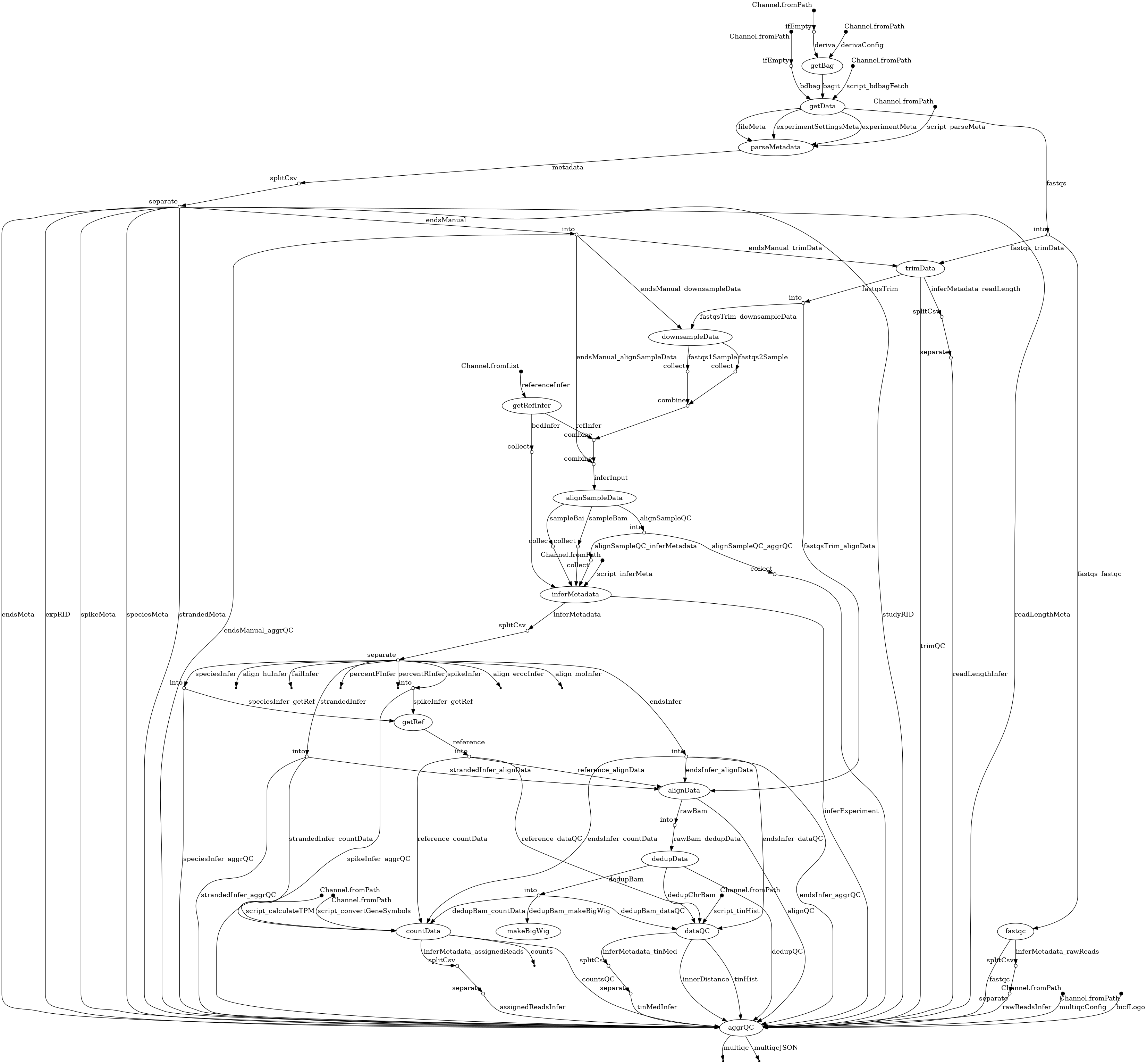
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- workflow/conf/multiqc_config.yaml 6 additions, 1 deletionworkflow/conf/multiqc_config.yaml
- workflow/rna-seq.nf 84 additions, 17 deletionsworkflow/rna-seq.nf
- workflow/scripts/convertGeneSymbols.R 24 additions, 0 deletionsworkflow/scripts/convertGeneSymbols.R
- workflow/scripts/parseMeta.py 5 additions, 0 deletionsworkflow/scripts/parseMeta.py
- workflow/scripts/splitStudy.py 24 additions, 0 deletionsworkflow/scripts/splitStudy.py
- workflow/scripts/splitStudy.sh 17 additions, 0 deletionsworkflow/scripts/splitStudy.sh
- workflow/scripts/tinHist.py 5 additions, 1 deletionworkflow/scripts/tinHist.py
- workflow/tests/test_consistency.py 1 addition, 1 deletionworkflow/tests/test_consistency.py
- workflow/tests/test_parseMetadata.py 1 addition, 1 deletionworkflow/tests/test_parseMetadata.py
{kind=link}
{kind=link}
| W: | H:
| W: | H:
workflow/scripts/convertGeneSymbols.R
0 → 100644
workflow/scripts/splitStudy.py
0 → 100644
workflow/scripts/splitStudy.sh
0 → 100644