This commit is part of merge request !43. Comments created here will be created in the context of that merge request.
Showing
- .gitlab-ci.yml 171 additions, 3 deletions.gitlab-ci.yml
- CHANGELOG.md 30 additions, 2 deletionsCHANGELOG.md
- README.md 7 additions, 0 deletionsREADME.md
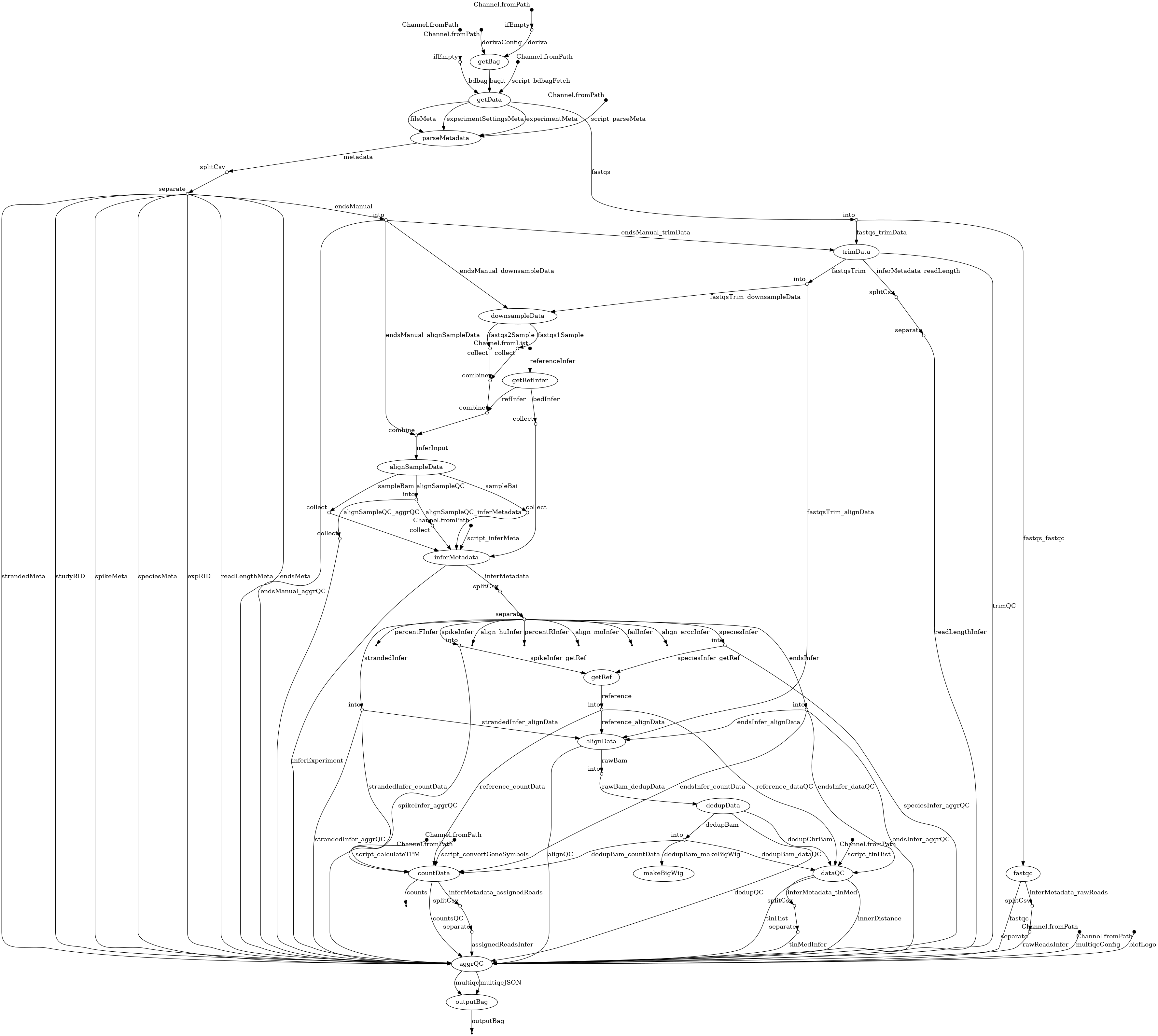
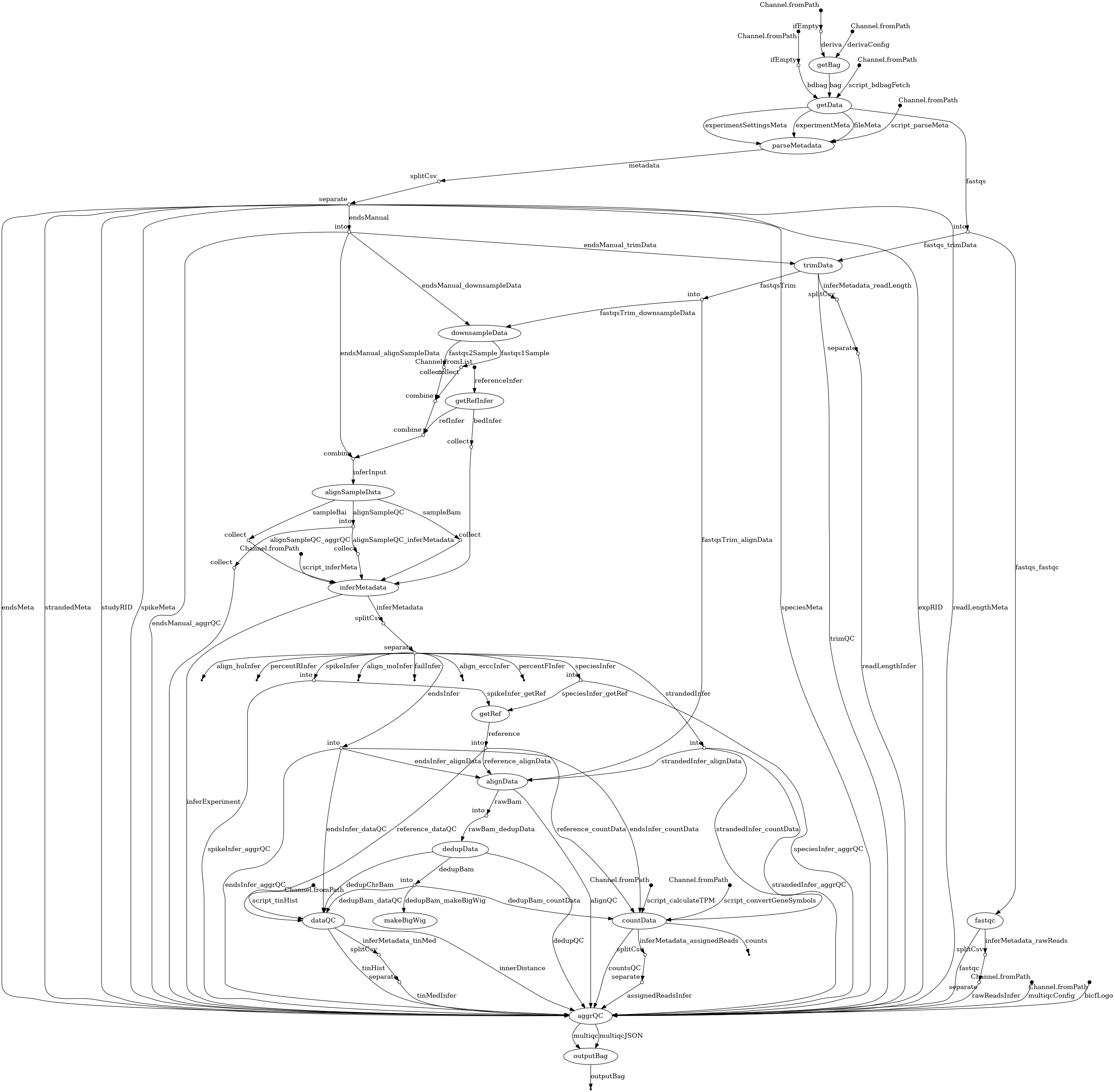
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- test_data/createTestData.sh 10 additions, 6 deletionstest_data/createTestData.sh
- workflow/conf/multiqc_config.yaml 43 additions, 2 deletionsworkflow/conf/multiqc_config.yaml
- workflow/nextflow.config 1 addition, 1 deletionworkflow/nextflow.config
- workflow/rna-seq.nf 104 additions, 23 deletionsworkflow/rna-seq.nf
- workflow/scripts/calculateTPM.R 5 additions, 2 deletionsworkflow/scripts/calculateTPM.R
- workflow/scripts/convertGeneSymbols.R 6 additions, 4 deletionsworkflow/scripts/convertGeneSymbols.R
- workflow/tests/test_makeFeatureCounts.py 2 additions, 1 deletionworkflow/tests/test_makeFeatureCounts.py
{kind=link}
{kind=link}
| W: | H:
| W: | H: