
Merge branch '78-tool_version' into develop
Showing
- .gitlab-ci.yml 100 additions, 0 deletions.gitlab-ci.yml
- .gitlab/merge_request_templates/Merge_Request.md 2 additions, 0 deletions.gitlab/merge_request_templates/Merge_Request.md
- CHANGELOG.md 1 addition, 0 deletionsCHANGELOG.md
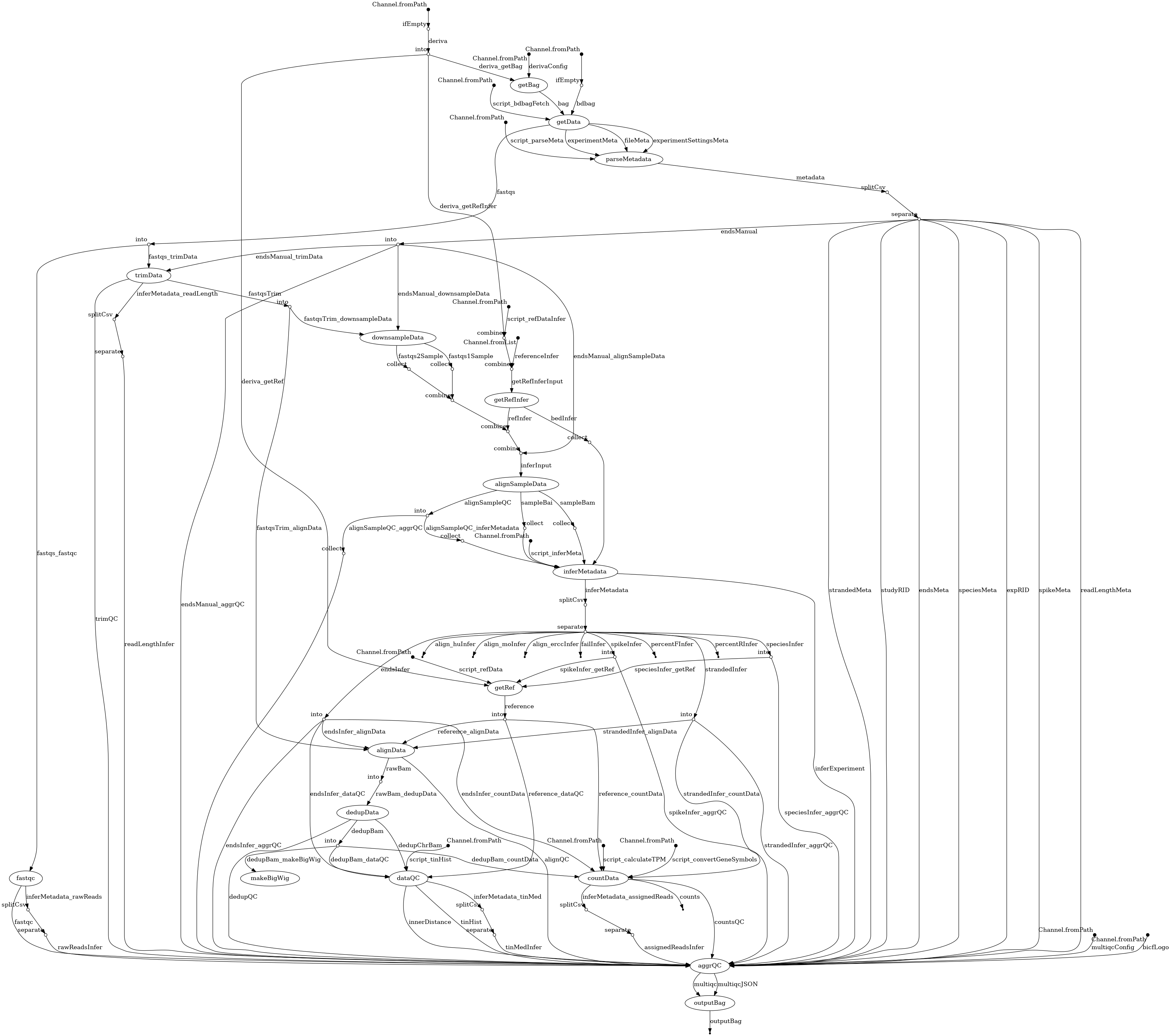
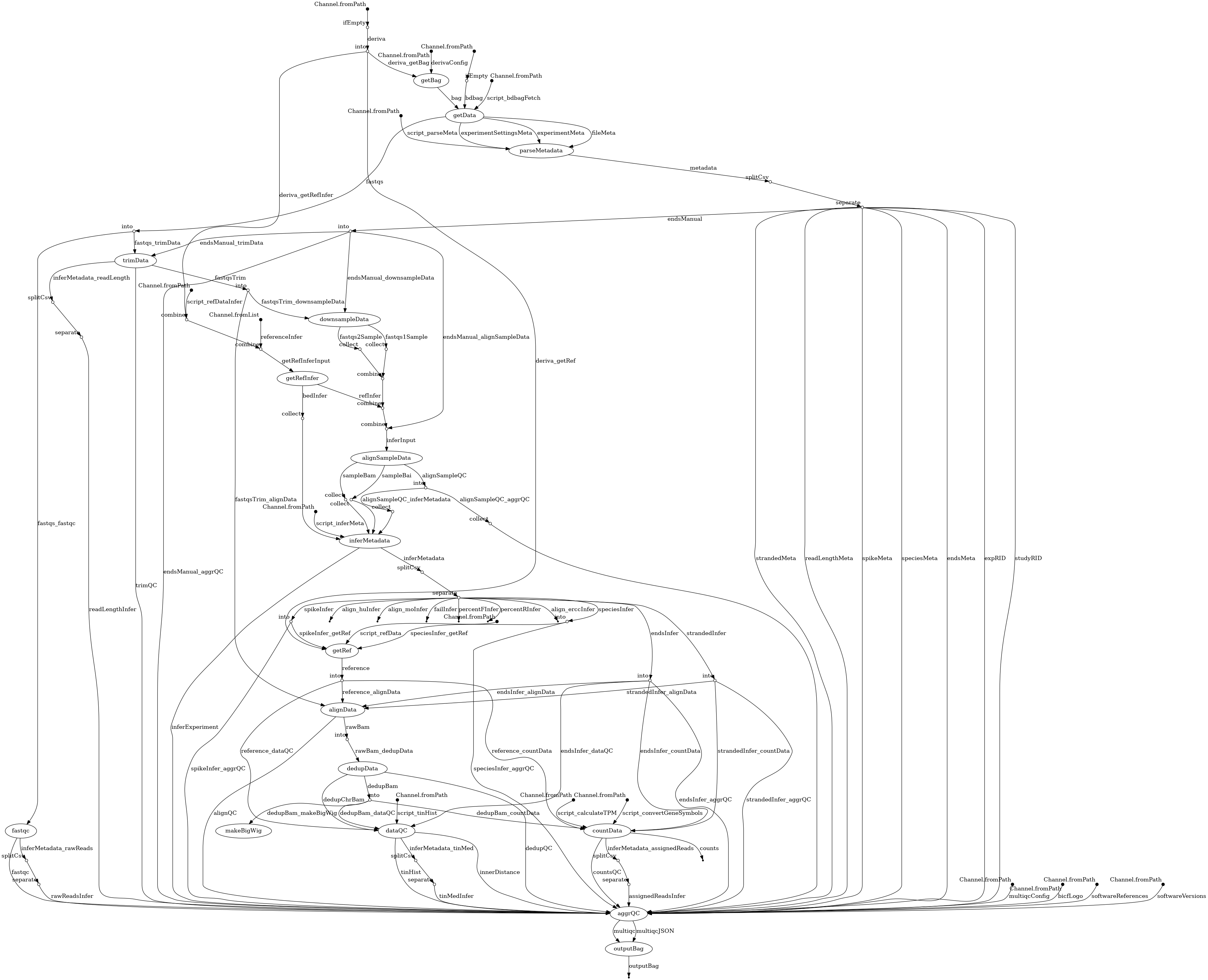
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- docs/references.md 42 additions, 0 deletionsdocs/references.md
- docs/software_references_mqc.yaml 93 additions, 0 deletionsdocs/software_references_mqc.yaml
- docs/software_versions_mqc.yaml 24 additions, 0 deletionsdocs/software_versions_mqc.yaml
- workflow/conf/multiqc_config.yaml 5 additions, 1 deletionworkflow/conf/multiqc_config.yaml
- workflow/rna-seq.nf 4 additions, 0 deletionsworkflow/rna-seq.nf
- workflow/scripts/generate_references.py 70 additions, 0 deletionsworkflow/scripts/generate_references.py
- workflow/scripts/generate_versions.py 142 additions, 0 deletionsworkflow/scripts/generate_versions.py
{kind=link}
{kind=link}
| W: | H:
| W: | H:
docs/software_references_mqc.yaml
0 → 100644
docs/software_versions_mqc.yaml
0 → 100644
workflow/scripts/generate_references.py
0 → 100644
workflow/scripts/generate_versions.py
0 → 100644