
Merge resolution
Showing
- .gitlab-ci.yml 14 additions, 8 deletions.gitlab-ci.yml
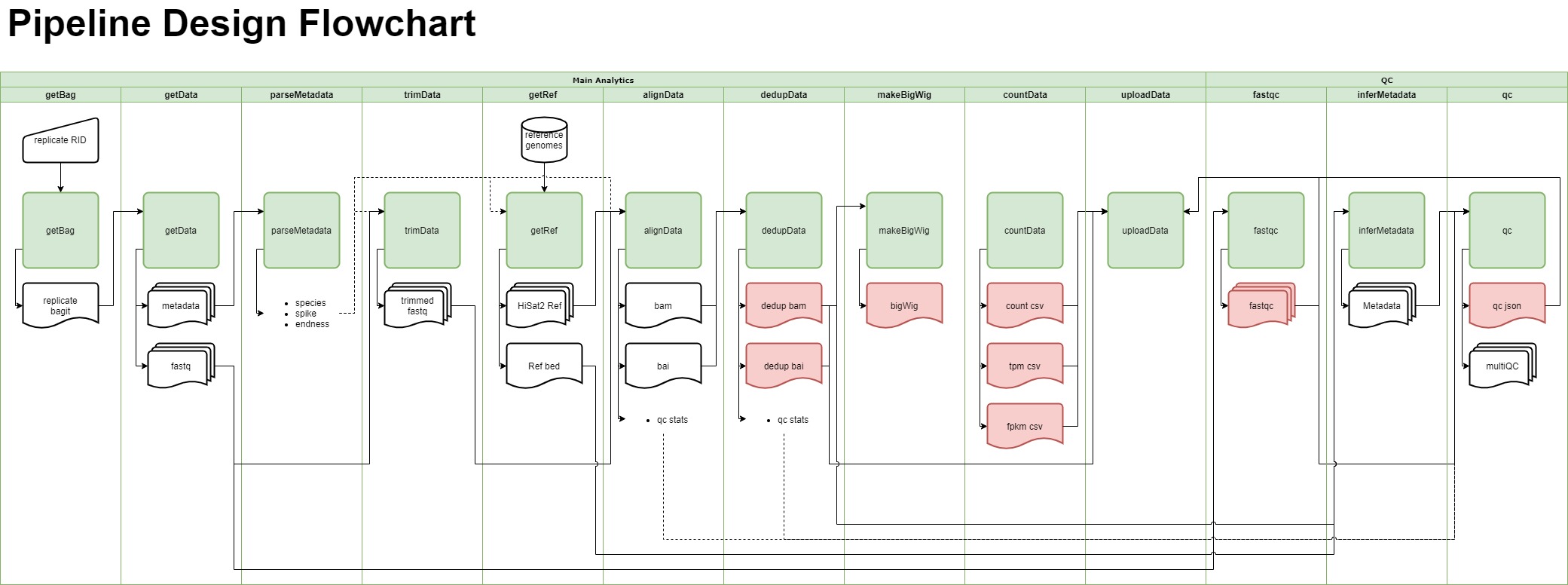
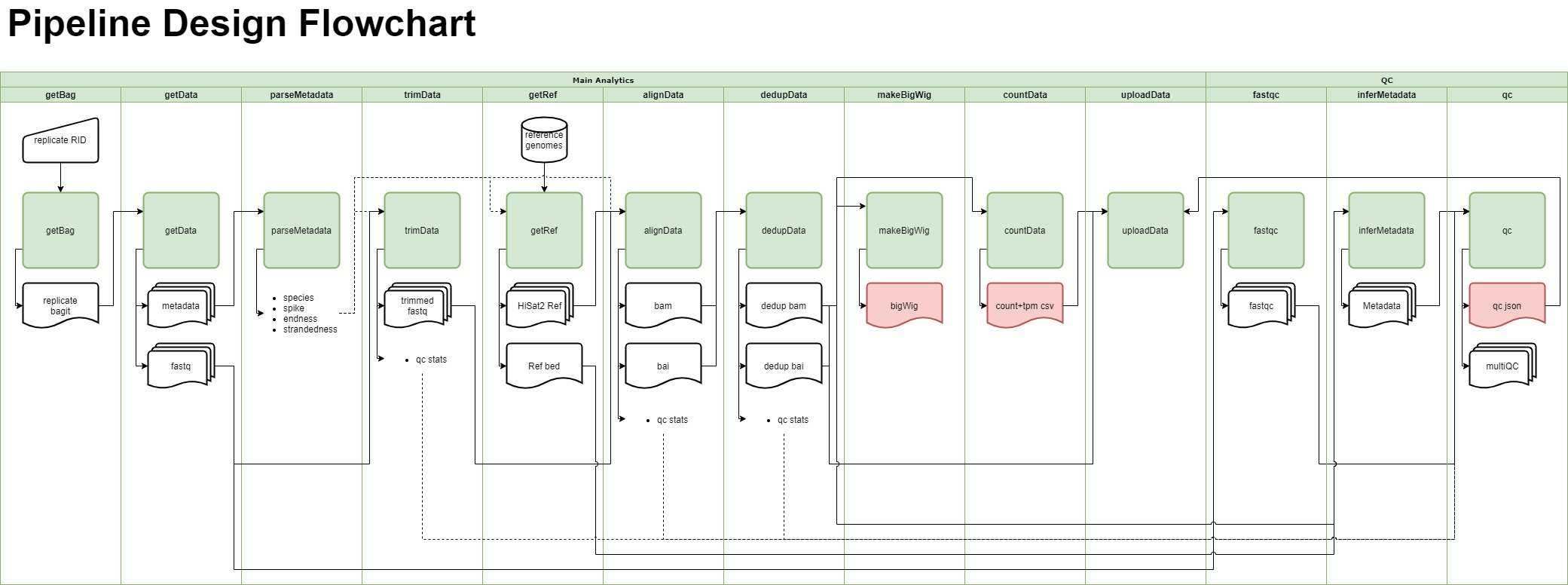
- docs/RNA-Seq Pipeline Design Flowchart.drawio 1 addition, 1 deletiondocs/RNA-Seq Pipeline Design Flowchart.drawio
- docs/RNA-Seq Pipeline Design Flowchart.jpg 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Flowchart.jpg
- docs/RNA-Seq Pipeline Design Flowchart.pdf 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Flowchart.pdf
- docs/RNA-Seq Pipeline Design Process Table.docx 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Process Table.docx
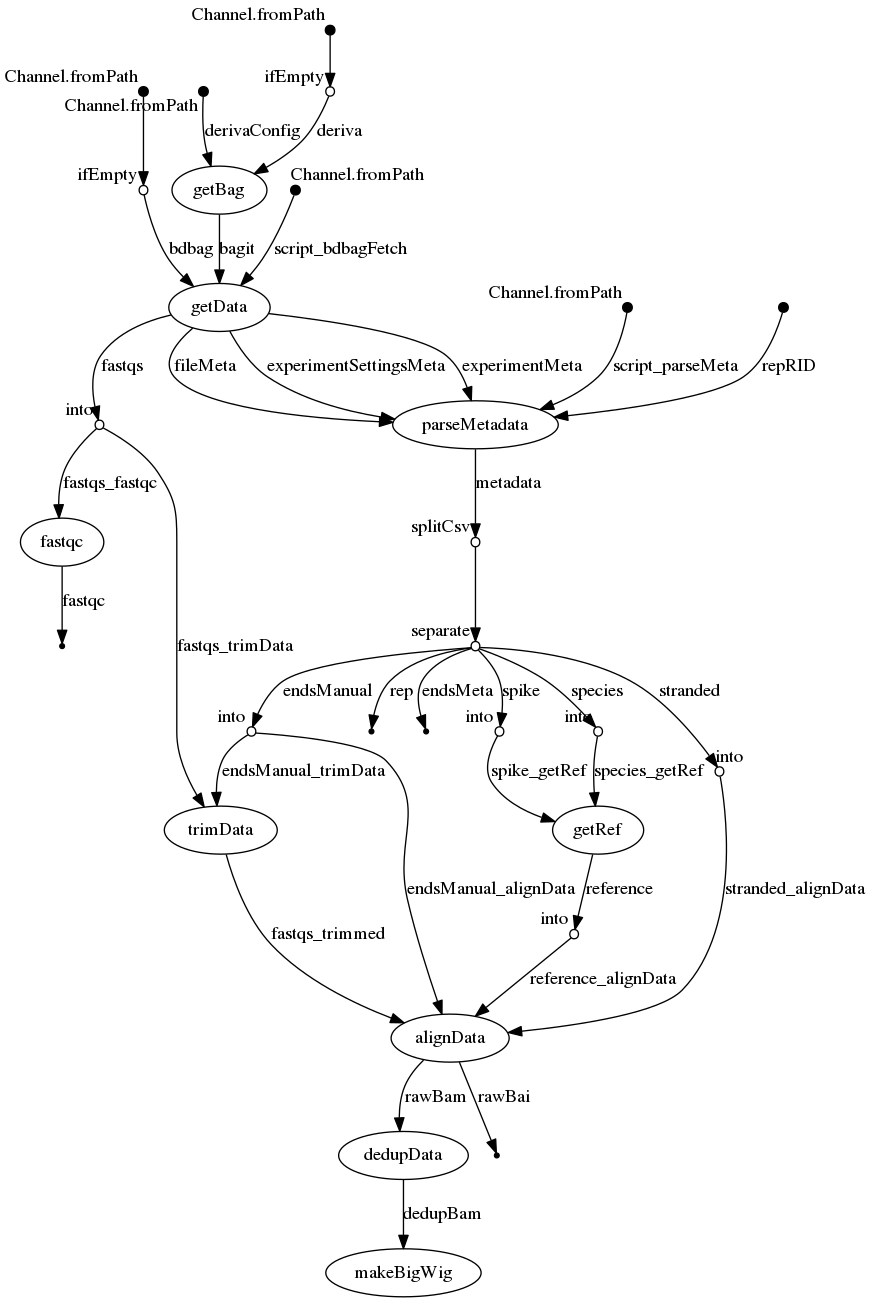
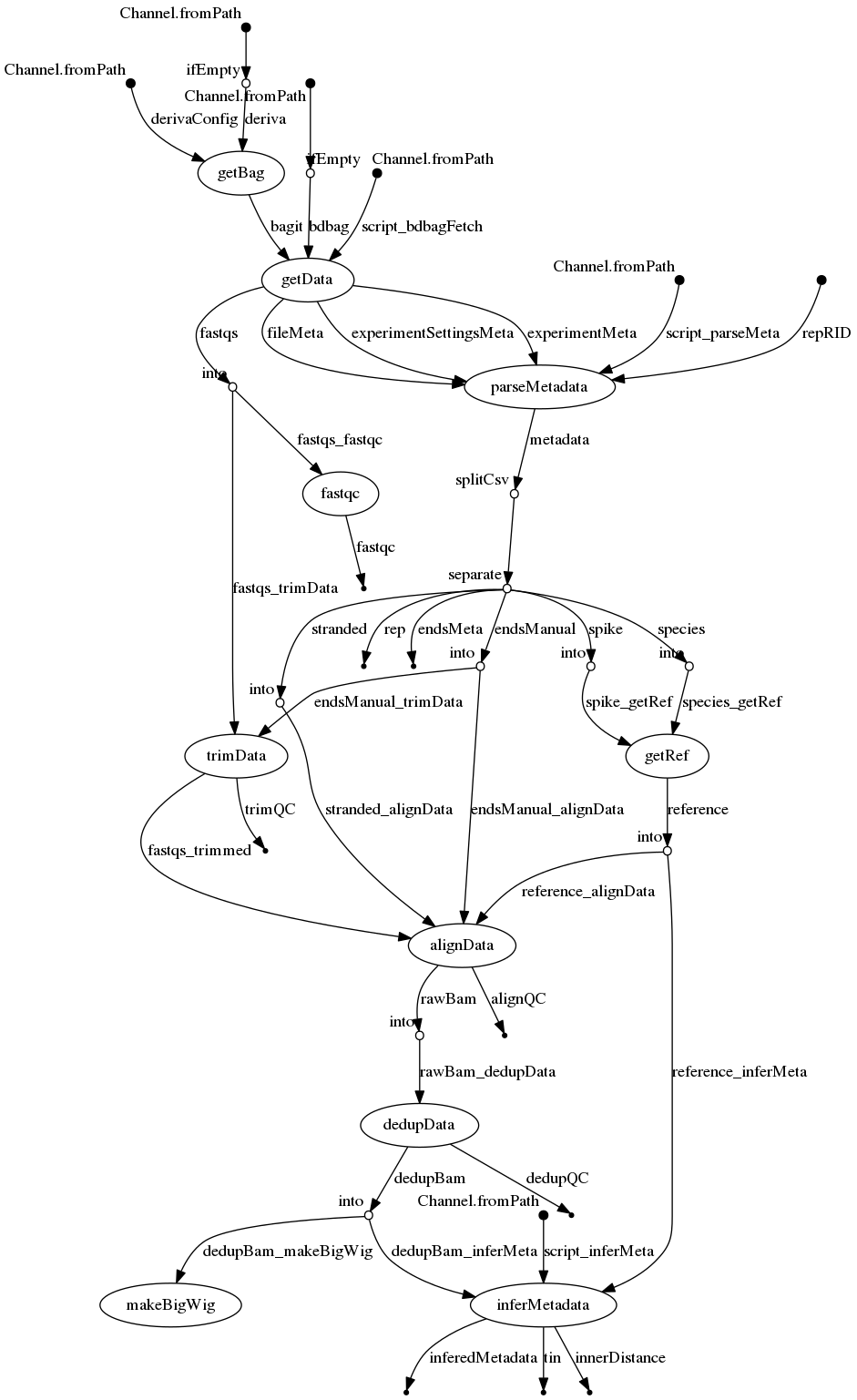
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- workflow/conf/biohpc.config 4 additions, 1 deletionworkflow/conf/biohpc.config
- workflow/nextflow.config 8 additions, 5 deletionsworkflow/nextflow.config
- workflow/rna-seq.nf 118 additions, 30 deletionsworkflow/rna-seq.nf
- workflow/scripts/bdbagFetch.sh 12 additions, 5 deletionsworkflow/scripts/bdbagFetch.sh
- workflow/scripts/inferMeta.sh 21 additions, 0 deletionsworkflow/scripts/inferMeta.sh
- workflow/tests/test_alignReads.py 2 additions, 2 deletionsworkflow/tests/test_alignReads.py
- workflow/tests/test_dedupReads.py 2 additions, 1 deletionworkflow/tests/test_dedupReads.py
- workflow/tests/test_getData.py 2 additions, 2 deletionsworkflow/tests/test_getData.py
- workflow/tests/test_inferMetadata.py 14 additions, 0 deletionsworkflow/tests/test_inferMetadata.py
{kind=link}
{kind=link}
| W: | H:
| W: | H:
File deleted
No preview for this file type
{kind=link}
{kind=link}
| W: | H:
| W: | H:
workflow/rna-seq.nf
100755 → 100644
workflow/scripts/inferMeta.sh
0 → 100644
workflow/tests/test_inferMetadata.py
0 → 100644