Merge branch '62-output.inputBagit' into 'develop'
Resolve "Add input bagit to ouput" Closes #62 See merge request !38
Showing
- .gitlab-ci.yml 10 additions, 4 deletions.gitlab-ci.yml
- CHANGELOG.md 5 additions, 0 deletionsCHANGELOG.md
- README.md 3 additions, 2 deletionsREADME.md
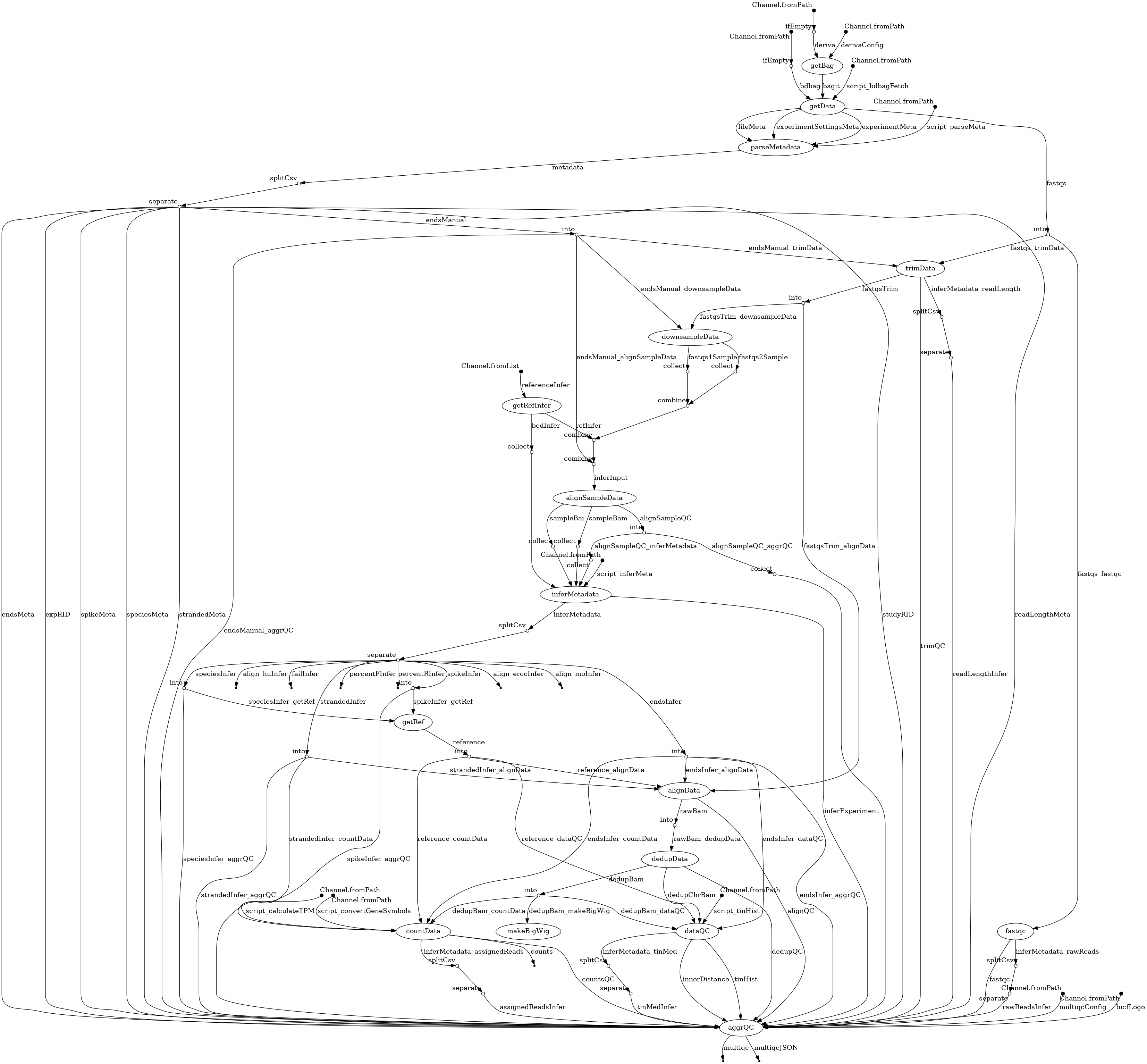
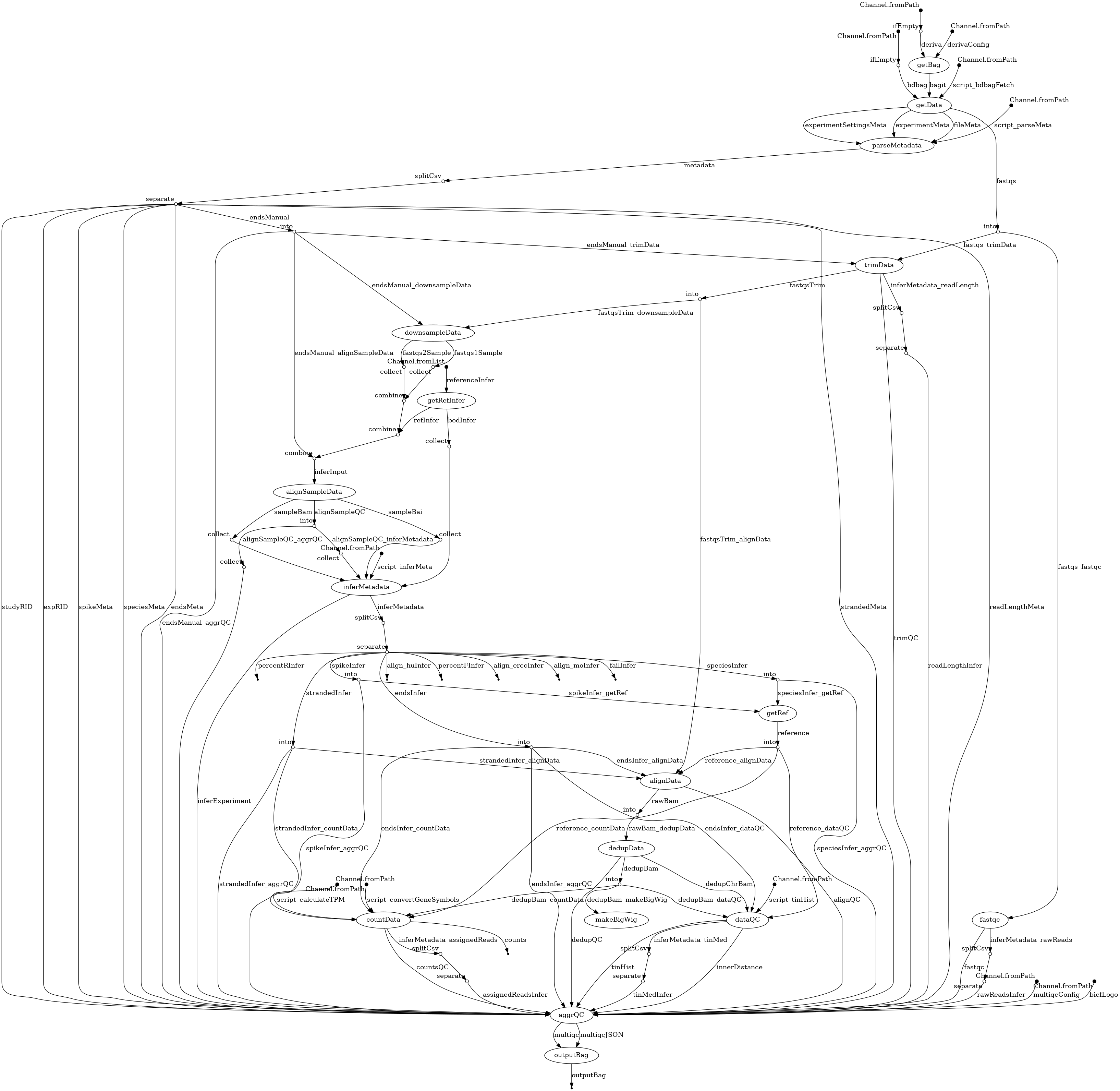
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- workflow/conf/aws.config 4 additions, 0 deletionsworkflow/conf/aws.config
- workflow/conf/biohpc.config 6 additions, 3 deletionsworkflow/conf/biohpc.config
- workflow/nextflow.config 7 additions, 4 deletionsworkflow/nextflow.config
- workflow/rna-seq.nf 29 additions, 4 deletionsworkflow/rna-seq.nf
- workflow/scripts/splitStudy.sh 5 additions, 1 deletionworkflow/scripts/splitStudy.sh
- workflow/tests/test_outputBag.py 13 additions, 0 deletionsworkflow/tests/test_outputBag.py
docs/dag.png
100644 → 100755
{kind=link}
{kind=link}
| W: | H:
| W: | H:
workflow/tests/test_outputBag.py
0 → 100644