There was an error fetching the commit references. Please try again later.
Merge
Showing
- .gitignore 2 additions, 1 deletion.gitignore
- .gitlab-ci.yml 515 additions, 78 deletions.gitlab-ci.yml
- .gitlab/merge_request_templates/Merge_Request.md 3 additions, 1 deletion.gitlab/merge_request_templates/Merge_Request.md
- CHANGELOG.md 54 additions, 0 deletionsCHANGELOG.md
- README.md 56 additions, 32 deletionsREADME.md
- docs/RNA-Seq Pipeline Design Flowchart.drawio 0 additions, 1 deletiondocs/RNA-Seq Pipeline Design Flowchart.drawio
- docs/RNA-Seq Pipeline Design Flowchart.jpg 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Flowchart.jpg
- docs/RNA-Seq Pipeline Design Process Table.docx 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Process Table.docx
- docs/RNA-Seq Pipeline Design Process Table.pdf 0 additions, 0 deletionsdocs/RNA-Seq Pipeline Design Process Table.pdf
- docs/bicf_logo.png 0 additions, 0 deletionsdocs/bicf_logo.png
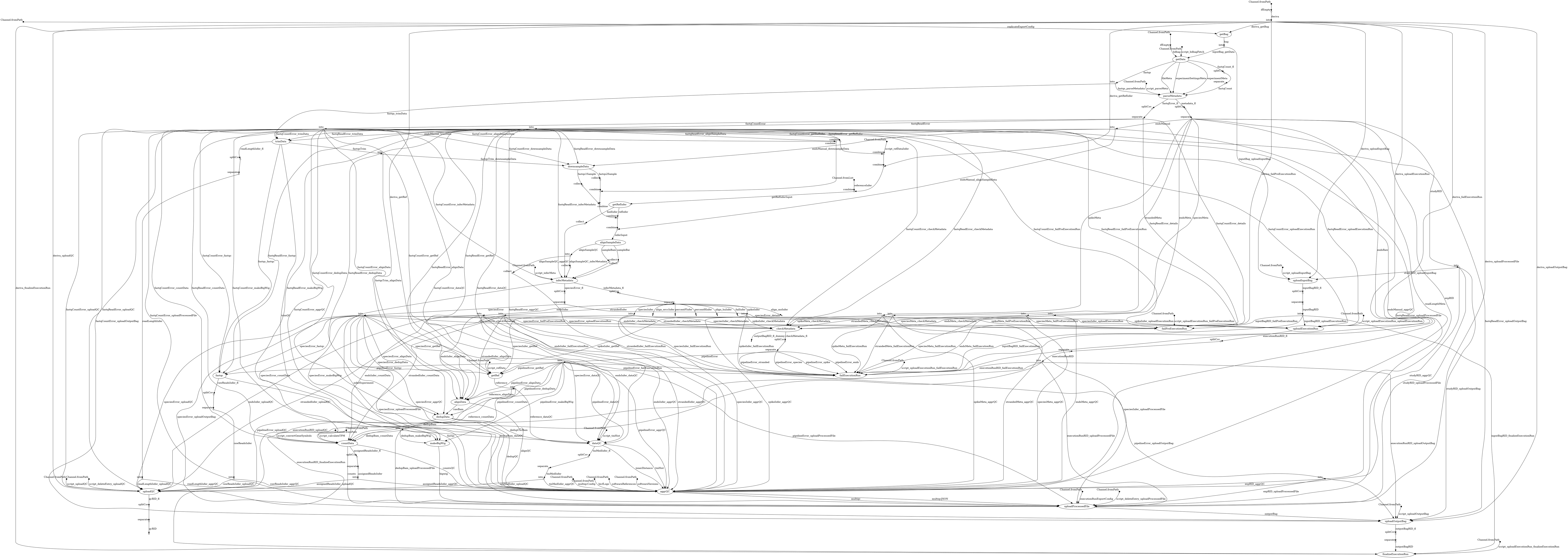
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- docs/references.md 42 additions, 0 deletionsdocs/references.md
- docs/software_references_mqc.yaml 93 additions, 0 deletionsdocs/software_references_mqc.yaml
- docs/software_versions_mqc.yaml 24 additions, 0 deletionsdocs/software_versions_mqc.yaml
- test_data/Replicate_For_Input_Bag(test).json 2 additions, 2 deletionstest_data/Replicate_For_Input_Bag(test).json
- test_data/createTestData.sh 43 additions, 38 deletionstest_data/createTestData.sh
- workflow/conf/Execution_Run_For_Output_Bag.json 64 additions, 0 deletionsworkflow/conf/Execution_Run_For_Output_Bag.json
- workflow/conf/Replicate_For_Input_Bag.json 97 additions, 0 deletionsworkflow/conf/Replicate_For_Input_Bag.json
- workflow/conf/aws.config 54 additions, 18 deletionsworkflow/conf/aws.config
- workflow/conf/biohpc.config 52 additions, 18 deletionsworkflow/conf/biohpc.config
This diff is collapsed.
{kind=link}

213 KiB
File deleted
File deleted
{kind=link}
{kind=link}
| W: | H:
| W: | H:
{kind=link}
{kind=link}

| W: | H:
| W: | H:
docs/software_references_mqc.yaml
0 → 100755
docs/software_versions_mqc.yaml
0 → 100755
workflow/conf/Replicate_For_Input_Bag.json
0 → 100644