There was an error fetching the commit references. Please try again later.
Merge branch '11-deriva.upload' into 'develop'
Resolve "process_derivaUpload" Closes #24, #75, and #11 See merge request !53
Showing
- .gitlab-ci.yml 291 additions, 69 deletions.gitlab-ci.yml
- .gitlab/merge_request_templates/Merge_Request.md 3 additions, 3 deletions.gitlab/merge_request_templates/Merge_Request.md
- CHANGELOG.md 8 additions, 3 deletionsCHANGELOG.md
- README.md 7 additions, 4 deletionsREADME.md
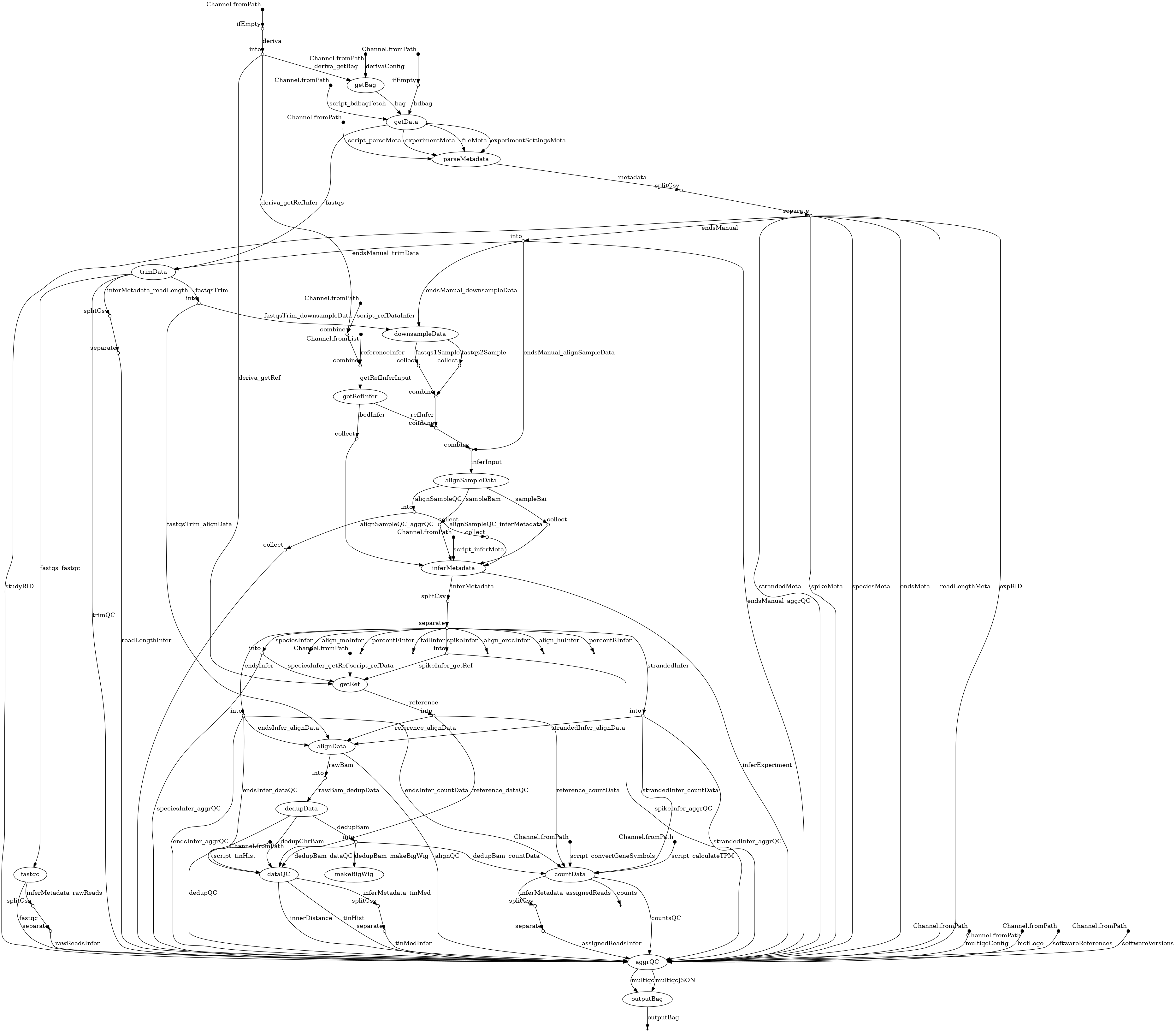
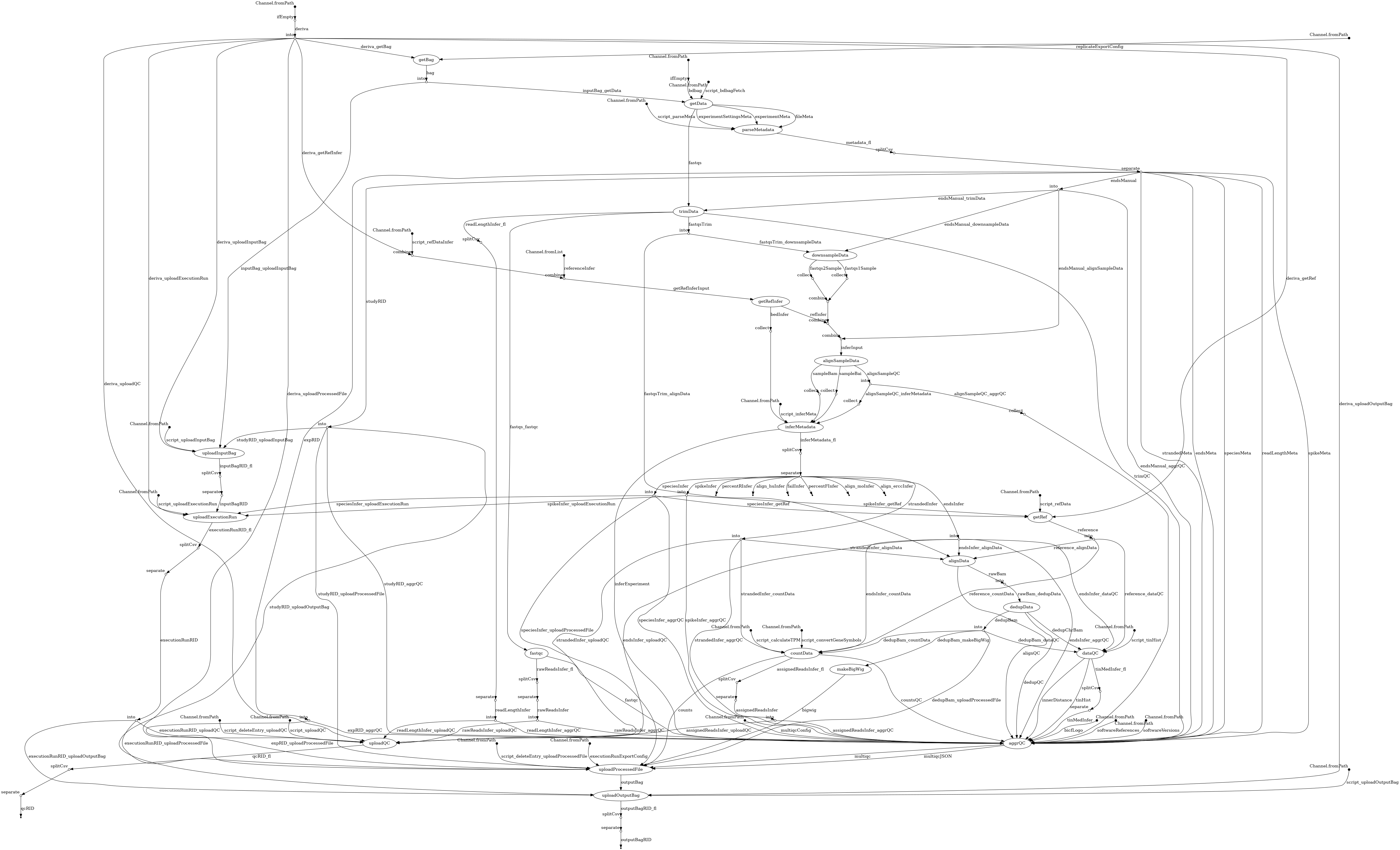
- docs/dag.png 0 additions, 0 deletionsdocs/dag.png
- docs/software_references_mqc.yaml 2 additions, 2 deletionsdocs/software_references_mqc.yaml
- docs/software_versions_mqc.yaml 12 additions, 12 deletionsdocs/software_versions_mqc.yaml
- test_data/createTestData.sh 41 additions, 38 deletionstest_data/createTestData.sh
- workflow/conf/Execution_Run_For_Output_Bag.json 64 additions, 0 deletionsworkflow/conf/Execution_Run_For_Output_Bag.json
- workflow/conf/Replicate_For_Input_Bag.json 1 addition, 1 deletionworkflow/conf/Replicate_For_Input_Bag.json
- workflow/conf/aws.config 17 additions, 1 deletionworkflow/conf/aws.config
- workflow/conf/biohpc.config 13 additions, 1 deletionworkflow/conf/biohpc.config
- workflow/conf/multiqc_config.yaml 55 additions, 34 deletionsworkflow/conf/multiqc_config.yaml
- workflow/nextflow.config 27 additions, 15 deletionsworkflow/nextflow.config
- workflow/rna-seq.nf 455 additions, 80 deletionsworkflow/rna-seq.nf
- workflow/scripts/bdbag_fetch.sh 0 additions, 0 deletionsworkflow/scripts/bdbag_fetch.sh
- workflow/scripts/convertGeneSymbols.R 1 addition, 1 deletionworkflow/scripts/convertGeneSymbols.R
- workflow/scripts/delete_entry.py 37 additions, 0 deletionsworkflow/scripts/delete_entry.py
- workflow/scripts/extract_ref_data.py 0 additions, 0 deletionsworkflow/scripts/extract_ref_data.py
- workflow/scripts/generate_versions.py 1 addition, 1 deletionworkflow/scripts/generate_versions.py
{kind=link}
{kind=link}
| W: | H:
| W: | H:
File moved
workflow/scripts/delete_entry.py
0 → 100644
File moved